Bulk RNA-seq, made approachable.
Pick a public dataset or upload your FASTQ. Describe what you're looking for in plain English. An hour later, you have a reproducible, sourced RNA-seq answer — DE tables, plots, QC reports — and an AI co-scientist that can read across runs, draft your Methods, and propose the next experiment.
Made in Europe, on European compute. Designed so biologists can drive their own analyses — and bioinformaticians can skip the boilerplate and focus on the interesting questions.
Drosophila, C. elegans, zebrafish. Mouse and human are deliberately out of scope for now — we'll open them when the clinical-grade governance is ready, not before.
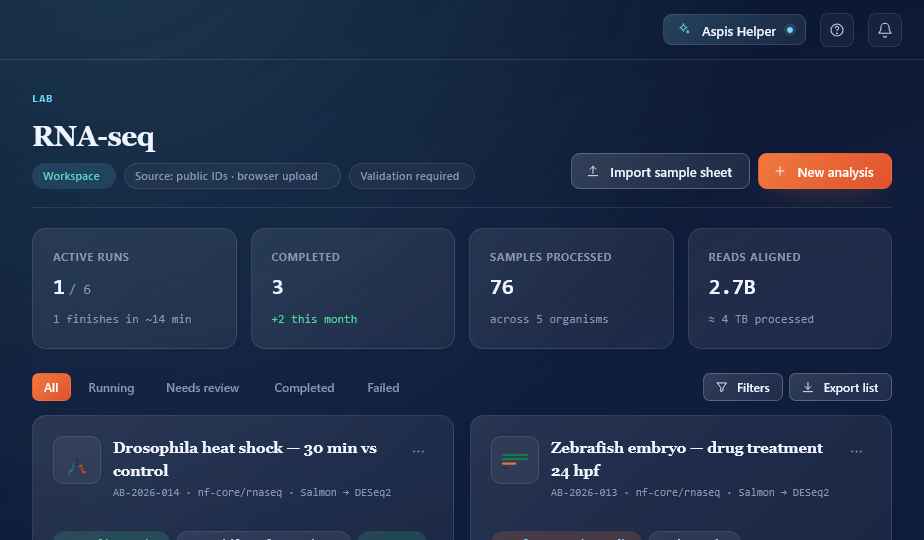
Everything in one project.
Samples, runs, QC, results, and the conversation with your AI co-scientist — all in one place. No more chasing logs across four tools and an email thread.
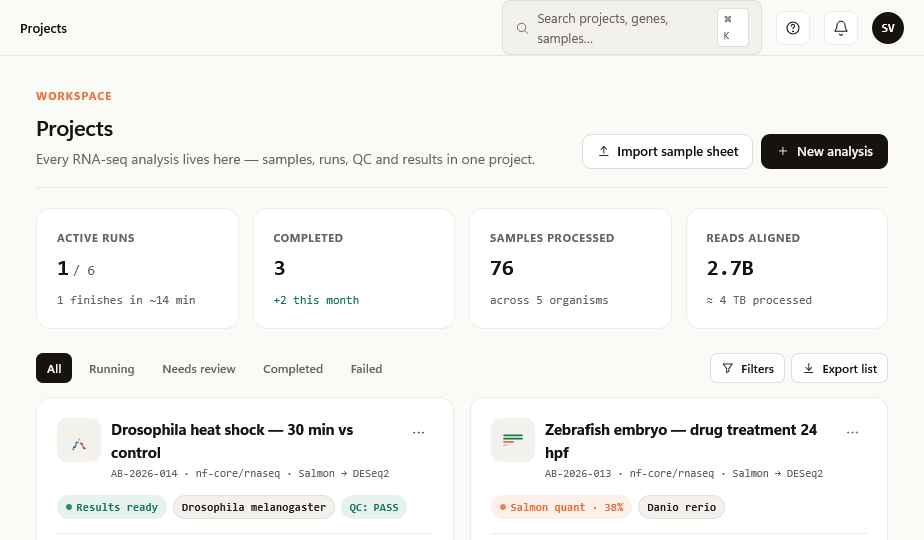
A single page for everything you need to look at.
Samples. Mapping rate. Up- and down-regulated genes. A clean catalog of every figure and table the run produced. One tab for the AI co-scientist. No tabs you've never used and never will.
Every number is traceable to a real artifact. Nothing on this page was invented — not by us, not by the AI.
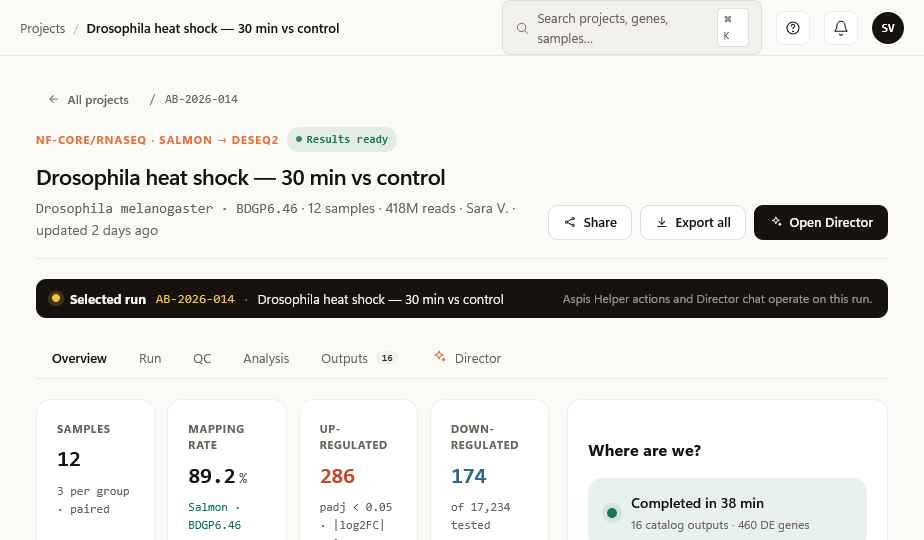
A co-scientist that reads your data.
Ask anything about your run — a gene, a contrast, a stuck step, a confusing QC warning. The Helper sees the run you have selected, looks at the actual outputs, and answers in plain English with the source attached.
It can validate your sample sheet before you submit. Explain why a step failed. Recommend the right contrast for your design. Draft the Methods paragraph when you're done.
Your raw data never enters the AI's context. It works on a sanitized summary — organism, samples, QC, available outputs — and the catalog of artifacts the run produced.
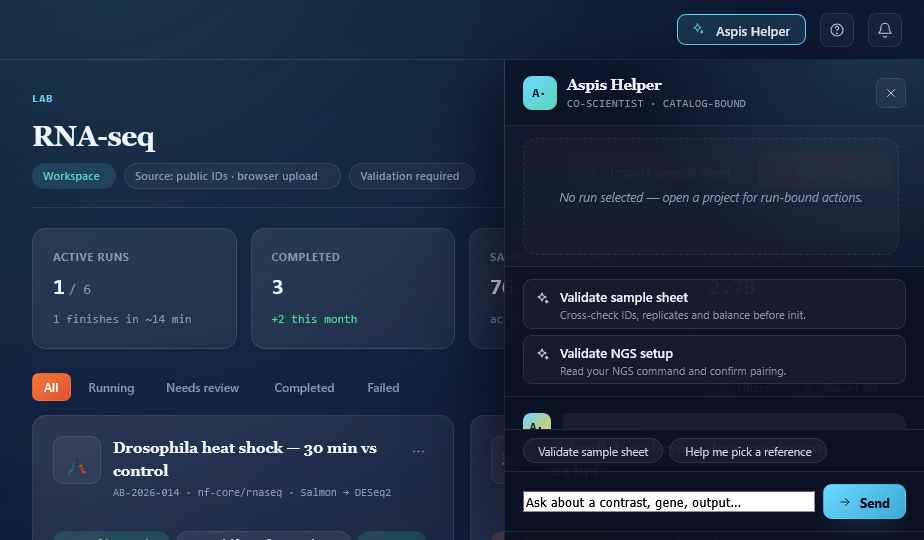
Beyond a single run.
Most of biology happens across runs, not inside one. Deep Analyze takes the runs you've completed, finds what they say together, and turns the answer into something you can put in a paper.
Compare
Pick two or more completed runs. See which genes overlap, which are run-specific, which directions agree or contradict. Drug treatment vs control across timepoints? Two genotypes against the same baseline? It's one click.
Report
"Draft me a Results paragraph for these three runs." The Helper writes a grant-ready or paper-ready section — with sample counts, mapping rates, the actual contrasts, the top genes, and a Methods block that matches the pipeline you used.
Hypotheses
Given what you found, what would you test next? The Helper proposes follow-up experiments — knockdowns, additional timepoints, rescue conditions — grounded in the actual genes and pathways your runs surfaced.
Interpret
Organism-specific biology with references. The Helper pulls from FlyBase, WormBase, ZFIN, KEGG, Gene Ontology, and the literature — every claim links to a real source. No invented papers, no fabricated gene functions.
Deep Analyze isn't isolated. It can pull readings from Biovision — your gel quantifications, your cell counts, your Western blots — and notes from Labbook, so your transcriptomic story sits next to the bench evidence that supports it. The same AI co-scientist, one continuous conversation across the entire lab.
Every plot, every table, ready when you are.
Count matrices, differential-expression tables, QC reports, PCA, volcano plots, MultiQC, heatmaps, deconvolution proportions. The standard outputs ship pre-computed; the rest render on demand when you ask for them.
Search by gene, contrast, file type, or scientific alias. Download what you want. Nothing in the picker accidentally triggers compute.
Bulk in, reference profiles out.
Aspis can run or stage reference-based deconvolution on bulk RNA-seq. Runnable references stay separated from imported-but-not-yet-buildable atlases, so the browser never presents a planned database as a finished matrix.
- FlyBase FCA full gene expression
- full FCA h5ad/loomX
- adult Drosophila midgut atlas
- WormSeq adult whole-body
- CeNGEN neuronal
- Cao/Shendure larval atlas
- CeSTAAN adult neuronal
- Daniocell/GSE223922 h5ad
- ZCL 1.0 scanpy h5ad
- ZCL raw DGE
- Daniocell Seurat v4
- Zebrahub
Runnable today means an implemented builder, exact source-file manifest gate, and runtime S3 placement exist. Candidate databases may be downloaded or manually exported into object storage, but they stay marked as import planned or manual export until a matrix materializer and validation gate are implemented.
Your runs, in your pocket.
Start a job from a public dataset. Watch mapping rate tick up over coffee. Get a push when DE results land. The same AI co-scientist, sized for one hand.
FASTQ files from your phone are intentionally disabled — keep big files on the workstation. The app is for starting public-dataset runs, monitoring jobs, and talking to the AI.
The pipeline is boring on purpose.
We use what the field already trusts. No bespoke aligners. No closed-source magic. Just a clean, container-pinned run on Scaleway, with every step logged and every artifact catalogued.
Three model organisms. Carefully.
Mouse and human references may exist in our object store but are product-blocked. We won't open them until the governance, consent, and clinical-grade audit story is built. AI free-form code execution is also disabled — the Director can only request whitelisted actions.
None of this exists without open source.
Aspis Bio RNA-seq is a thin, careful layer on top of decades of community work. Every tool below is maintained by researchers who chose to share their code with the field. Thank you.
Join the waitlist for RNA-seq.
RNA-seq rolls out as part of the Aspis Bio alpha. Drop your email and we'll let you know when your spot is ready.